The Truth About Serious Lung Diseases: Causes, Risks & Prevention Tips
Your lungs are vital organs that keep your body supplied with oxygen and free from harmful gases. Yet, millions of people worldwide suffer from serious lung diseases that can drastically impact quality of life—and even become life-threatening. From chronic conditions to infections, understanding the causes, risks, and prevention strategies is key to protecting your respiratory health.
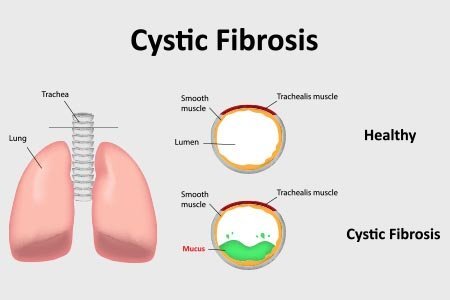
What Are Serious Lung Diseases?
Serious lung diseases are conditions that affect the structure or function of the lungs, making it difficult to breathe properly. Some of the most common and dangerous ones include:
These conditions can range from manageable chronic illnesses to severe, life-threatening diseases requiring urgent medical care.
Common Causes of Lung Diseases
Understanding the root causes can help you reduce your risk:
1. Smoking and Tobacco Use
Smoking is the leading cause of lung diseases, especially COPD and lung cancer. Even secondhand smoke can damage lung tissues over time.
2. Air Pollution
Exposure to polluted air, especially in urban environments, increases the risk of respiratory conditions. Fine particles and toxic gases can inflame and damage the lungs.
3. Infections
Bacterial, viral, and fungal infections can severely affect lung health. Diseases like tuberculosis and pneumonia can cause long-term complications if untreated.
4. Occupational Hazards
People working in industries with dust, chemicals, or fumes are at higher risk. Long-term exposure can lead to chronic lung conditions.
5. Genetic Factors
Some lung diseases, such as certain types of pulmonary fibrosis, may have a genetic link.
Key Risk Factors You Should Know
Certain lifestyle and environmental factors can increase your chances of developing serious lung diseases:
- Long-term smoking or vaping
- Living in highly polluted areas
- Weak immune system
- Lack of physical activity
- Poor nutrition
- Family history of lung disease
Recognizing these risk factors early can help you take preventive action.
Warning Signs of Lung Disease
Many lung diseases develop slowly and silently. Watch out for these symptoms:
- Persistent cough
- Shortness of breath
- Chest pain or tightness
- Wheezing
- Coughing up blood
- Fatigue and weakness
If you experience any of these symptoms, consult a healthcare professional promptly.
Why Early Detection Matters
Early diagnosis can significantly improve treatment outcomes. Regular health check-ups and screenings are especially important if you are at high risk. Conditions like lung cancer, when detected early, have a much higher survival rate.
Internal Resource for Better Health
For more insights on protecting your overall health, don’t miss our detailed guide:
👉 Love Your Liver: Natural Ways to Keep Your Liver Strong & Prevent Liver Disease
Taking care of your liver and lungs together ensures a stronger, healthier body.
Conclusion
Serious lung diseases are a growing health concern, but they are often preventable with the right knowledge and lifestyle choices. By understanding the causes, recognizing the risks, and following simple prevention tips, you can safeguard your lung health for years to come.
Remember: every breath you take is precious—protect your lungs today for a healthier tomorrow.
References:
https://www.mayoclinic.org/diseases-conditions/copd/symptoms-causes/syc-20353679
https://my.clevelandclinic.org/health/diseases/4375-lung-cancer
https://www.webmd.com/asthma/what-is-asthma/
https://www.medicinenet.com/pulmonary_fibrosis/article.htm
Medications that have been suggested by doctors worldwide are available on the link below
https://mygenericpharmacy.com/category/disease/asthma-allergies