Important treatment option to consider for Cystic Fibrosis.
A hereditary disorder that affects the lungs, digestive system, and other organs is called cystic fibrosis (CF). When the protein responsible for producing mucus does not function normally, it develops. As a result, the body produces thick, sticky mucus that can harm or clog organs.
Although there is no known cure for the condition, medicines can help control it, ease the symptoms, and lower the chance of consequences. As a result, a person’s life expectancy and quality of life are improved.
What is Cystic fibrosis?
The lungs and digestive system are the two main organs affected by the hereditary illness CF. Additionally, it may result in consequences including diabetes and liver disease. A gene termed the cystic fibrosis transmembrane conductance regulator has a hereditary mutation in CF patients (CFTR). The CFTR protein is regulated by this gene.
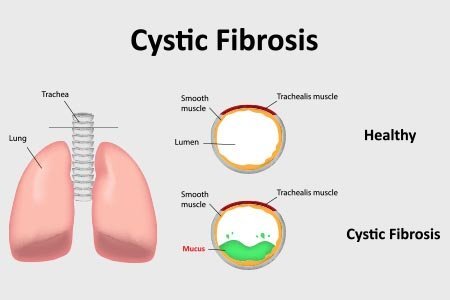
Every organ that produces mucus contains the protein. Additionally, various tissues and organs, such as those in the:
- lungs
- pancreas
- intestines
- liver
- heart
- system of defence
- sweat ducts
The CFTR protein does not operate as it would in a healthy organism due to the genetic mutation. The body produces mucus that is thicker and stickier than usual as a result of this dysfunction. It may close off the airways, resulting in serious lung infections and breathing problems.
The pancreatic enzymes may not be able to adequately break down food as a result of the genetic mutation, which could further affect pancreatic function. Digestion issues may result from this and result in stunted growth and malnutrition.
A chronic illness, CF can have complications that could be fatal. Treatments, however, can lengthen and improve the quality of life.
Symptoms of Cystic fibrosis.
Most frequently, CF affects the lungs, leading to symptoms of the respiratory system like:
- wheezing
- breathing difficulty
- a prolonged cough that occasionally produces blood or mucous
- further breathing problems
Additionally, the mucus that prevents lung function in CF patients provides ideal living conditions for infections. A person is therefore at a higher risk of developing lung infections including bronchitis and pneumonia.
CF symptoms might differ from person to person based on the organs that are impacted. Other potential signs and issues include:
- sinus infections frequently
- gastrointestinal conditions like:
- abdomen ache
- constipation
- diarrhoea
- oily, unpleasant stool
- Small, fleshy growths inside the nose called nasal polyps
- perspiration and salted skin
- morning sweats
- fever
- jaundice
- muscle and joint ache
- low body mass
- little development or weight increase in children
- postponed puberty
- infertility in men
- Due to a shortage of oxygen reaching the extremities, people have clubbed fingers and toes.
In addition to increasing the risk of diabetes and osteoporosis, pancreatic blockage can result in malnutrition and stunted growth.
Causes of cystic fibrosis
The “cystic fibrosis transmembrane conductance regulator” gene, often known as the CFTR gene, is the cause of CF. This gene regulates the flow of salt and water into and out of the cells in your body.
Your mucus becomes thicker and stickier than it should be as a result of a sudden mutation, or alteration, in the CFTR gene. Your perspiration contains more salt as a result of this abnormal mucus, which also accumulates in a number of body organs, including the:
- intestines
- pancreas
- liver
- lungs
The CFTR gene is susceptible to a variety of abnormalities. The severity of CF is correlated with the kind of defect. The child inherits the faulty gene from their parents.
Cystic Fibrosis Diagnosis
Early detection translates into quicker treatment and greater long-term health. Every state in the United States uses one or more of these three tests to check neonates for cystic fibrosis:
- Blood test. This examination measures the amount of immunoreactive trypsinogen (IRT). Blood levels of it are higher in those with CF.
- DNA analysis. This checks for CFTR gene mutations.
- Sweat test. Your sweat’s salt content is measured. Results that are higher than usual point to CF.
Some infants who weren’t screened for CF aren’t given the diagnosis until they are adults. If you exhibit symptoms of the disease, your doctor could do DNA or sweat testing on you.
Cystic fibrosis (CF) treatment
Your medical team will likely include a cystic fibrosis expert in addition to many other kinds of providers. Although there is no treatment for cystic fibrosis, your team will assist you in managing the condition. Keeping your airways open is the main goal of management. When necessary, your doctor will also write prescriptions for medication.
Clearing the airways
If you have cystic fibrosis, you can maintain your airways open in a number of ways:
- You can pick up different breathing and coughing techniques.
- You can employ treatment vests that use vibrations to loosen mucus or mouthpiece devices.
Chest physical therapy, also known as postural drainage and percussion to remove mucus, is a skill you may master. This technique involves moving into specific positions to allow your lungs to empty. To help loosen the mucus, another person may place their touch on your back or chest. You might cough while doing this.
Cystic fibrosis medications
These drugs, which won’t treat CF but will be helpful to you in some circumstances, may be prescribed by your doctor. They consist of:
- Antibiotics to treat or prevent lung infections.
- Breathing is made easier by inhaling bronchodilators, which widen and relax your airways.
- medication was inhaled to thin the mucus and make it easier to expel.
- steroids and non-steroidal anti-inflammatories, which are anti-inflammatory medications.
- medications for those with specific gene variations to treat the underlying causes of cystic fibrosis.
- Digestive enzymes from the pancreas.
- Stool softeners for constipation relief.
Cystic fibrosis operations
Surgery can be required if you have cystic fibrosis or one of its side effects.
These could consist of:
- surgery on the sinuses or nose.
- bowel surgery to clear obstructions.
- procedure involving organ transplantation, such as a liver or double lung transplant.
When to see a doctor
Consult your doctor about getting tested for the disease if you or your kid exhibit symptoms of cystic fibrosis or if someone in your family has the condition. Consult a medical professional who is familiar with CF.
Your doctor must be followed up with consistently and regularly, at least every three months, if you have cystic fibrosis. If you notice any new or worsening symptoms, such as more mucus or a change in the colour of your mucus, fatigue, loss of weight, or severe constipation, speak to your doctor right once.
If you experience severe stomach discomfort and distention, chest pain, difficulty breathing, or are coughing up blood, get immediate medical attention.
REFERENCES:
- https://www.healthline.com/health/cystic-fibrosis
- https://www.medicalnewstoday.com/articles/147960
- https://www.mayoclinic.org/diseases-conditions/cystic-fibrosis/symptoms-causes/syc-20353700
- https://my.clevelandclinic.org/health/diseases/9358-cystic-fibrosis
- https://www.webmd.com/children/what-is-cystic-fibrosis
For more details, kinlty visit below.